
There are millions of volunteers who take part in clinical trials every single year. Before participating in any government- or industry-sponsored research, every subject has the right to be informed about what will happen next and how it will happen. Such process in which subject A is provided with the necessary details by subject B is called informed consent. But let’s view this from a slightly larger and more thorough prism. Different trials, of course, carry different risks and/or benefits.
Full comprehension of all procedures during the trial is mandatory because only this way the person can adequately acknowledge advantageous or disadvantageous aspects like these in order to decide whether or not his/her participation is the best choice for him/her. It is the staff duty to let one individual know the ins and outs of a specific clinical study but it is a responsibility of the individual to ask further questions when one thing or another seems vaguely explained, unclear or even disturbing. In other words, the informed consent is all about clarification.
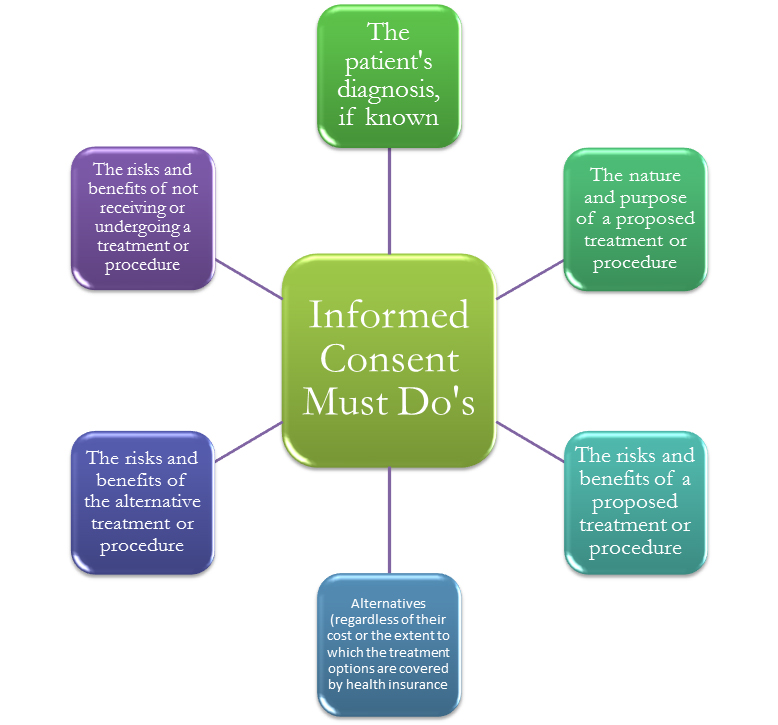
In clinical trials we have informed consent document and informed consent process. The first one offers a summarized version of the procedure. The consent document reveals the purpose of the trial, the treatment course of action and schedule, eventual risks, benefits and other possible treatments apart from the one being offered. The informed consent process, on the other hand, gives ongoing explanations to the subject which will help him/her decide whether or not to start or keep on with the participation in the trial.
Nevertheless, there has been a significant change in the practice of consent taking across time. If in the present day it is regarded as a Good Clinical Practice (GCP) requirement which exhibits legal and moral characteristics, in the past the value of consent taking was, in one way or another, disregarded and even nonexistent. Going back to the Second World War, we can find plenty of cases which disclose research atrocities. Back then doctors based their practices on nothing else but deception and concealment of information from the patient. The number of non-specified diagnostic and ambiguous instructions made the doctor-patient relationship paternalistic. In this regard, an example of unethical doctors’ behavior and clinical procedures is the experiments of Nazi medics on prisoners in concentration camps. Most of the conducted researches included deliberate shooting of captivated people so that doctors could study gunshot injuries; intentional infection of wounds in order to try out new antimicrobial products and so on.
Fortunately, in the twenty first century, following informed consents is a must and failure to comply with the preset standards and requirements can have severe consequences such as disciplinary action by the actual professional authority, criminal action or civil litigation. With the shift of the understanding of human rights, we can observe various new trends and aspects in the nature of the informed consent in clinical trials.
- Disclosure
Prior to any official start of a clinical research, complete transparency is required. Hence, the doctor is obliged to discuss any potential risks with the patient. In addition to this, physicians are also supposed to disclose other likely treatment options, uncertainties, benefits and risks in case treatment is sought.
- Patient’s understanding
The comprehension of the patient is another core factor when obtaining an informed consent. The staff is expected to approach patients in the most appropriate way so that they can have enough time to digest what is being explained to them and to fully understand the unveiled particularities regarding the following procedure. If interpreters are used, the doctor must make sure that the interpreter has presented every detail to the patient correctly and hasn’t omitted any relative information.
- Competence
All patients who give informed consent are expected to be competent to make the decision on their own. Here competence is determined as the capacity of the participant to give consent. Patient’s capacity rests in the legal age of majority.
- Mental capacity
Another cardinal aspect is for the patient to be able to make adequate decisions when giving their consent. To ensure the patient’s mental condition, a mental capacity assessment may be necessary to carried out. In cases when patients cannot make decisions on their own, a substitute decision maker (usually a parent or another relative) is allowed to give the consent.
- Autonomy
Something else that has been circling around the newly arisen trends which have an impact on the informed consent is the autonomy of patients. In this sense their agreement to proceed with a certain treatment or other type of medical procedure should be entirely voluntary and no third parties have affected their final decision.
For the last couple of years there have been noted some quite remarkable tendencies in the practice of informed consent in clinical trials. With paramount aspects such as disclosure of information, patient’s understanding, autonomy, competence and mental capacity, giving consent transforms into a process with far more strict requirements than in the past. Failure to obtain proper informed consent which meets all of the predetermined conditions will result in serious penalties. If you are interested in knowing more, we have an open training available to anyone on Informed Consent, registration is free here.
PDF Version of this article available here: https://crotraining.co.uk/wp-content/uploads/2015/05/Informed-Consent-in-Clinical-Trials-New-Trends-and-Aspects.pdf